J. Phys. B: At. Mol. Opt. Phys. 31 (1998) L103–L107. Printed in the UK LETTER TO THE EDITOR The binding of positronium to lithium
Faculty of Science, Northern Territory University, Casuarina, NT, 0909, Australia
Abstract.
The wavefunction of the lithium positride (LiPs) ground state is calculated using
an ab initio approach which uses a Gaussian basis with r2 correlation factors. The energy of
the LiPs ground state, −7.738 55 Hartree, has a total energy more negative than the sum of thepositronium (−0.250 Hartree) and lithium (−7.478 04 Hartree) ground states. The ground stateis therefore stable against dissociation into Li + Ps with a binding energy of −0.010 51 Hartree.
The use of positrons as a tool for spectroscopy, for example, positron annihilationspectroscopy in condensed matter physics, has gained widespread acceptance over the years[1, 2]. One of the questions that is raised by the use of positrons as a spectroscopic tool is ofcourse the possibility that chemically stable systems containing a positron, or positronium,could be formed in the various targets under investigation [1, 2]. In spite of much work(and a lot of speculation), little hard information is known about the ability of a positronor positronium to form chemically stable systems in atoms, molecules or condensed matter. For instance, a recent review identified only five atoms and molecules (this excludes Ps−and Ps2) as permitting positronium binding with any degree of certainty [3]. These speciesare H, F, Cl, Br and OH. Most of the evidence for binding is based on calculation, and onlyfor one species, namely H, could the result be classed as rigorous. While the most recentresults for the other species come from high-quality calculations [4–6], they are based onmodel potentials for open-shell systems and do not have the same degree of certainty asthe result for hydrogen which has been the subject of numerous high-quality calculations[7–11] and a recent experiment [12].
Recently, the e+–Li species has been shown to be chemically stable with a binding
= 0.002 17 Hartree [13]. This result, and the fact the PsH is chemically
stable, immediately suggests that lithium positride, LiPs, would also be chemically stable. However, two earlier calculations on LiPs had failed to show that positronium could bebound to neutral lithium [14, 15]. Fortunately, we were ignorant of these results whendeciding whether to investigate the LiPs species for possible binding.
The condition for the LiPs ground state to be chemically stable is that the total energy of
the LiPs ground state be more negative than the sum (−7.728 04 Hartree) of the Ps groundstate energy (−0.250 Hartree) and the Li ground state energy (−7.478 04 Hartree) [13, 16]. The decay into Li− + e+ does not enter into the picture since the binding energy of the Li−ground state [13, 17] is only −7.500 76 Hartree.
In this letter, a large variational calculation of the LiPs system is performed using the
stochastic variational method (SVM) as implemented by Varga and Suzuki [18, 19]. TheSVM was initially proposed as a method suitable for solving nuclear structure problems
involving a small number of particles [20, 21]. The SVM uses a Gaussian basis whichhas a number of features that make it possible to generate very accurate wavefunctionsfor few-body systems.
First, the matrix elements of the interaction Hamiltonian can
be calculated analytically, or at worst, reduced to a one-dimensional integral for anynumber of particles.
Second, the Gaussian basis contains terms with r2 correlation
factors. In addition, that part of the wavefunction concerned with the spatial coordinatesmaintains its functional form after any possible permutation of the particles.
a very useful property for studying systems containing identical particles.
important aspects of any variational calculation is the optimization of the nonlinearparameters. In the SVM the exponents of the Gaussian basis are optimized using stochastictechniques.
In recent years, the SVM and related methods have been used to perform high-precision
variational calculations in atomic, mesoatomic, hypernuclear and multiquark systems [11,22–24]. The program of Varga and Suzuki [18] was used for the present set of calculations.
An initial series of calculations on a variety of related species were performed to validate
the method. Results of our calculations for Li+, Li, Li− and PsH are shown in table 1 andcompared with other accurate nonrelativistic calculations. To simplify comparison withother results in table 1, all results were computed with an infinite nuclear mass.
calculation for positronium hydride, PsH, agrees with the best previous calculation to within4 × 10−6 Hartree [11]. Results for Li− underestimate the best calculation [17] by less than7.0 × 10−4 Hartree. Table 1. Non-relativistic energies (in Hartree) of various atomic systems compared with previous accurate results. In these calculations the nuclear mass has been assumed to be infinite. The number in parentheses refers to the total dimension of the Gaussian basis.
a Reference [25]. b Reference [11]. c Reference [16]. d Reference [17].
The convergence of energy of the LiPs system as a function of the dimension of the
Gaussian basis is shown in table 2. In contradiction with previous works [14, 15] it isfound that the ground state is chemically stable. It proved relatively easy to demonstratethat the system was stable since binding was achieved with less than 200 basis functions. The largest calculation included 600 basis functions, and resulted in a total energy ofE = −7.738 55 Hartree which is equivalent to a binding energy of
When reference is made to the binding energy of the LiPs system it should be noted that thebinding energy is relative to breakup into Li and Ps. It is clear from the convergence patternshown in table 2 that the present energy does not represent the converged limit for the LiPstotal energy. This is not surprising since there are five active particles, four electrons andone positron in the LiPs system. We suspect that the present estimate of the binding energyis accurate to about 10%.
Convergence of the LiPs energy (in Hartree) as a function of basis size.
third column shows the binding energy (in Hartree) relative to the Li + Ps threshold at
Energy expectation values were also computed with the present optimized wavefunctions
for a finite mass. The 7Li nucleus has a mass of M = 12 863.2me and for this species weobtained E = −7.477 40 Hartree for Li, and E = −7.737 91 Hartree for LiPs giving abinding energy of
= 0.010 51 Hartree. Finite-mass considerations have almost no effect
on whether the system will be bound or not. Relativistic effects will also not affect whetherbinding is possible or not since one estimate of the relativistic energy correction for neutralLi is 0.000 011 Hartree [26].
Although the best ab initio estimate of the LiPs binding energy, 0.010 51 Hartree,
is subject to uncertainties due to incomplete convergence of the LiPs energy, and lessimportantly subject to relativistic and finite mass corrections, the statement that the systemis chemically stable will remain valid under any possible refinements of the model.
While the LiPs ground state is chemically stable, it is not stable against electron–positron
annihilation. The dominant decay process for electron–positron annihilation is into two γ -rays. The two-photon annihilation rate
2γ was computed using the general formula [11],
2γ = π α4c δ /a0
= 50.308 740 45 × 109 δ s−1.
In the above expression, δ is the expectation value of electron–positron Dirac δ-function,
δ(ri − r0)
where r0 is the positron coordinate and the ri (i = 1, 2, 3, 4) are the electron coordinates. The value obtained for the LiPs annihilation rate,
annihilation rate for PsH was also computed for validation reasons and the value weobtained,
= 2.45 × 109 s−1, was consistent with the best previous estimate [11],
2γ = 2.437 × 109 s−1.
The probability density for finding an electron or positron as a function of the distance
from the nucleus is depicted in figure 1. P−(r) is the electron probability density and P+(r)is the positron probability density. These functions are normalized so that
P+(r) dr = 1.
The electron density exhibits the shell structure of the atom with the probability densitiesfor the core and valence electrons having two distinct maxima. The peak of the probability
Figure 1. The electron and positron probability densities, P±(r), plotted as a function of r (in units of a0). The electron density is represented by the full curve while the positron density is shown by the broken curve.
density for the valence electrons occurs near 3.6 a0. The peak of the probability densityfor the positron occurs near 5.2 a0. That the positron orbits at a larger distance than theelectrons is expected since the electrons are attracted to the nucleus, while the positron isrepelled from the nucleus.
The binding of positronium to lithium immediately raises the question as to whether it is
possible to bind positronium to the heavier alkali atoms such as sodium and potassium. Thisquestion cannot be directly answered by SVM calculations since a full n-body calculationon a system containing 12 electrons and one positron (for sodium) is out of the question. The complexity of the problem needs to be reduced by using the frozen-core approximationbefore progress can be made.
The authors would like to thank Dr D M Schrader for useful correspondence and Dr K Vargafor the use of the SVM program. This work was supported by a research grant from theAustralian Research Council. References
[1] Schrader D M and Y C Jean (eds) 1988 Positron and Positronium Chemistry (Amsterdam: Elsevier)[2] Schrader D M and Wedlich D M 1989 From Atoms to Polymers: Isoelectronic Analogies ed J F Liebman
[3] Schrader D M Bound states of positrons with atoms and molecules: theory Nucl. Instrum. Methods under
[4] Schrader D M, Yoshida T and Iguchi K 1993 J. Chem. Phys. 98 7183 [5] Schrader D M, Yoshida T and Iguchi K 1992 Phys. Rev. Lett. 68 3281 [6] Yoshida T, Miyako G, Jiang N and Schrader D M 1996 Phys. Rev. A 54 964 [7] Ore A 1951 Phys. Rev. 83 665 [8] Lebeda C F and Schrader D M 1969 Phys. Rev. 178 24 [9] Clary D C 1976 J. Phys. B: At. Mol. Phys. 9 3115
[10] Ho Y K 1986 Phys. Rev. A 34 609 [11] Frolov A M and Smith V H Jr 1997 Phys. Rev. A 55 2662 [12] Schrader D M, Jacobsen F M, Frandsen N-P and Mikkelsen U 1992 Phys. Rev. Lett. 69 77 [13] Ryzhikh G G and Mitroy J 1997 Phys. Rev. Lett. 79 4124
[14] Harju A, Barbiellini B and Nieminen R M 1996 Phys. Rev. A 54 4849 [15] Saito S L 1995 Chem. Phys. Lett. 245 54 [16] McKenzie D K and Drake G W F 1991 Phys. Rev. A 44 R6973 [17] Fischer C F 1993 J. Phys. B: At. Mol. Opt. Phys. 26 855 [18] Varga K and Suzuki Y 1998 Comput. Phys. Commun. 106 157 [19] Varga K and Suzuki Y 1995 Phys. Rev. C 52 2885 [20] Kukulin V I 1975 Izv. Acad. Nauk 39 535 [21] Kukulin V I and Krasnopolsky V M 1977 J. Phys. G: Nucl. Phys. 3 795 [22] Kopylov V A, Frolov A M and Kolesnikov N N 1984 Izv. Vuzov 46 122 [23] Kolesnikov N N and Tarasov V I 1982 Sov. J. Nucl. Phys. 35 354 [24] Barbour I M and Ponting D K 1979 Nucl. Phys. 149 534 [25] Pekeris C L 1958 Phys. Rev. 112 1649 [26] Davidson E R, Hagstrom S A, Chakaravorty S J, Umar V M and Froese Fischer C 1991 Phys. Rev. A 44
TriCor Technologies Terms and Conditions of Sale These TriCor Technologies Terms and Conditions of Sale ("Agreement") apply to all quotations made by and purchase orders entered into by TriCor Technologies for the sale of standard and custom goods and services (collectively, "Product" or "Products") to Customer. Additional Terms and Conditions apply for t
My research interests focus on understanding, at a biochemical and structural level, the mechanisms that drive molecular machines. In particular my aim is to use cryo-EM in combination with image processing techniques to uncover conformational changes that play functional roles in protein translation (ribosome) and polyketide synthesis (PKS). Principle Investigator, CIC bioGUNE, Bilbao, Spain.
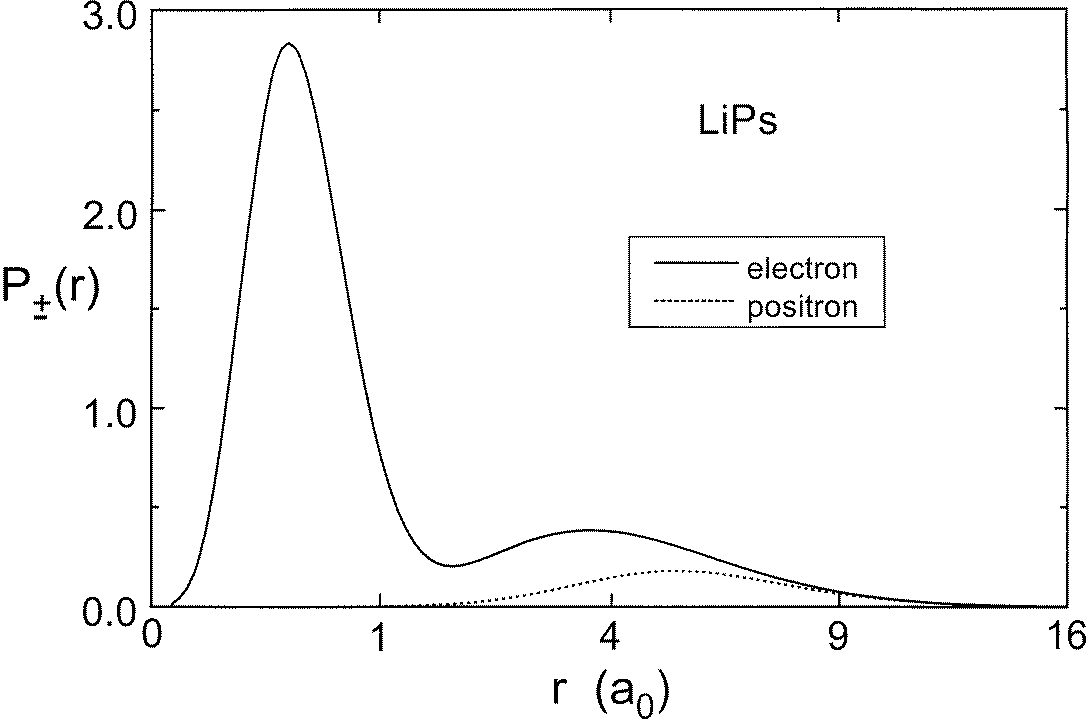 Figure 1. The electron and positron probability densities, P±(r), plotted as a function of r (in
Figure 1. The electron and positron probability densities, P±(r), plotted as a function of r (in