Contents
Solvent extraction is a technique which has been highly developed within many nationalnuclear energy programs because of its suitability as a selective separation process for fissionproducts, actinides, and other radioactive substances. The technique is briefly described inseveral sections of this book (§§ 5.5.3, 9.2.6, 9.4.3, 15.7.4, 16.3.3, 21.7, and especially in21.6 on the Purex process). It is based on the formation of uncharged organic metal complexeswhich are preferentially soluble in organic solvents. The three main types of such compoundsare: (i) organic chelate (i.e. binding through two positions in the organic molecule, like a claw)complexes such as plutonium tetraacetylacetonate, PuAa (general formula MA ), §9.4.3, cf.
Figs. 9.3, 9.9. and 9.11; (ii) inorganic metal complexes forming adducts with solvating organic compounds like TBPand hexone, e.g.UO (NO ) )(TBP) (or generally ML S , where L is an inorganic anion and
S the organic adduct former, §21.6, cf. Fig. 16.8); (iii) ion pair complexes between large organic cations (like alkylammonium ions, R NH+) and
negatively charge inorganic complexes like UO (SO )4 (generally RB
The physical and chemical principles governing the formation of extractable metal complexes,the conclusions which can be drawn about the chemical system from solvent extraction studies,and analytical and industrial applications are described in several monographs, e.g. "Principlesand Practices of Solvent Extraction" (1992). A.1. Single stage batch extractions
Let us consider a solute (e.g. uranium) which is distributed between an organic and anaqueous phase, independent of the kind of compounds the solute forms in the two phases. Afterequilibrium the weight of solute in the organic phase (extract) is w and in the aqueous phase
FIG. A.1. Different types of continuous extraction equipment. (a) Mixer-settler. (b) Spray column. (c) Pulsed column. (d) Tubular centrifugal contactor.
The weight fraction in the organic phase is
where the index 1 refers to the conditions after one extraction. P is called the extraction (orpartition) factor and defined by
where D is the distribution factor, defined by
Concentration of all M-species in the organic phase
D = ________________________________________________________
Concentration of all M-species in the aqueous phase
Radiochemistry and Nuclear Chemistry
and 2 is the phase volume ratio v /v . The percentage solute extracted, denoted E(%), is
equal to 100 R . The fraction of nonextracted solute left in the organic phase is n , and R +
Assume that one wants to separate uranium from the fission product lanthanum using thetwo-phase system: aqueous 1 M HNO , the organic adduct former tributyl phosphate, TBP
(100%, i.e. undiluted). In this system D = 20 for U(VI), while for lanthanum D = 0.07.
If one extracts with a phase ratio of 0.5, then R = 0.909 (E = 100 R = 90.9%) and R
= 0.034 (E 3.4%). This may be unsatisfactory with respect to both uranium yield and purity.
The yield can be increased by repeated extractions of the same aqueous phase (multipleextraction with one stationary phase, or "crosscurrent extraction"). For n such extractions onefinds that
Suppose n = 3 for our example, then n = 0.00075, i.e. for the three organic phase volumes
taken together E = 100(1 ! n ) = 99.92%. However, for lanthanum n
E = 9.8%. Although the uranium yield is high, the lanthanum impurity may be intolerable.
A more elaborate technique must be employed in order to obtain both high yield and highpurity under such conditions. Many such batch laboratory techniques have been described usingalternatively fresh organic (extraction) and aqueous (washing) solutions. The extractions arecarried out in special multistage equipment (§A.4). A.2. Multiple stage continuous processes
A single partitioning of a compound between an organic solvent and water may not besufficient for isolating it in acceptably pure form and good yield. Various multiple extractiontechniques may therefore be required. Such techniques have been described in §21.6.4 and theirtechnical application for uranium production (§5.5.3) and spent fuel reprocessing (§21.6.3). Continuous processes are preferred in industry, where the most common and simple solventextraction equipment is the mixer-settler (Fig. A.1(a); cf also Fig. 5.2). This type of equipmentis also becoming standard in laboratories engaged in process development. In the uraniumindustry a single mixer-settler may hold as much as 1000 m3. The mixer-settlers, each closelycorresponding to a single ideal extraction stage, are arranged in batteries containing any numberof stages. In these batteries the aqueous and organic phases flow countercurrent to each other(see Fig. A.2). For countercurrent solvent extraction, either batch or continuously, one finds that for thestationary state and for n stages
FIG. A.2. Mixer-settler countercurrent solvent extraction battery.
provided P is constant through all stages. In our example one obtains for 2 = 0.5 (flow rateratio organic : aqueous) and n = 3 that E = 99.91% and E = 3.5%. Thus about the same
yield of uranium, but with a somewhat lower lanthanum impurity as compared to thecrosscurrent extraction procedure. This impurity figure can be lowered substantially bymodification of the extraction process according to Figure A.3 so that the extraction batterycontains n extraction stages with extraction factor P , and m ! 1 washing stages with the
n = ______________________________________________________
which in the case that P = P reduces to
In the latter case, using three extraction stages and one washing stage, we find for ourexample that E is 99.9% and E is 0.12%. Thus both high yield and high purity are
FIG. A.3. Countercurrent solvent extraction with n extraction and m washing stages. Radiochemistry and Nuclear Chemistry
FIG. A.4. Notations for counter current extraction with wash stages
achieved with the countercurrent, central or intermediate feed solvent extraction technique. Thisis extensively used in uranium production, nuclear fuel reprocessing, and transuranium elementseparations. When the conditions are selected so that a high (Po1) extraction factor is obtainedfor the desired product and low ones (Pn1) for the impurities, high purity and good yield canbe obtained with relatively few stages. However, 2 is always lower in the extraction stages thanin the washing stages because the central feed flow rate adds to the wash flow rate in theextraction stages and the organic flow rate is unchanged. Hence, cases with the same P-valuein extraction and washing stages seldom occur. The purpose of the washing (or scrubbing) stages is to clean the desired product fromimpurities, and should not be confused with the stripping stages, the purpose of which is totransfer the product from the organic phase to a new aqueous phase. However, stripping canalso be so arranged that further purification is achieved, sometimes even using a central organicfeed, cf. Fig. A.4. If no additional purification is desired after extraction and the extractant isvolatile, the organic phase may be distilled, leaving a pure solid product. A.3. High loadings
A further industrial requirement on solvent extraction processes is high capacity, which meanshigh concentrations of several solutes in the aqueous and organic solvents ("high loading"). Theextraction factor P (or distribution factor D ) may then vary from stage to stage because D-
value and phase volumes change. The calculation of the number of stages,
flow rate ratios, etc., needed in order to obtain the desired product has to take this into account. In order to treat industrial conditions with coupled extraction and washing batteries withcentral feed where both phase volumes and distribution ratios may change as an effect of theextraction we introduce the mass flow rates: > = XV and 0 = YV , where X and Y are the
concentrations in the aqueous and organic phases respectively, and V and V are the flow
rates of the aqueous and organic phases. We also redefine the extraction factor for the j:thstage, P , in terms of actual flow rates out of that stage as
where the index j+1 means that it flows to the next higher stage and j that it flows to theprevious stage, see Figure A.4. Now five useful extraction functions can be defined as follows.
Through a series of mass balances we can obtain the following equation for mass flow rate withthe outgoing aqueous phase, >P
> = [> + 0 (e . + s ) + > s + 0 s ]/(e . + s )
The symbols refer to those given above and in Figure A.4. Mass flow rates and conditionsinside each battery can in principle be obtained recursively from the relations
Radiochemistry and Nuclear Chemistry
However, numerical problems tend to make the use of these equations difficult for batterieswith many stages. Special cases occur when P either is constant or constantly equal 1. The flow rates out from any stage can usually be calculated with sufficient accuracy from theapparent molar volumes, M , of the i compounds according to
3(1 + M X ) and V = V
are the flow rates of the pure unloaded phases in each stage. In many
cases one of the central feeds is missing (usually the organic feed) and the corresponding massand volume flow is zero. The wash stream (index W) is then the real feed for that phase. When the flow rates vary considerably in the batteries due to loading effects, the wholecalculational procedure becomes recursive and a computer code based on these equations maybe needed. However, in most cases it is only necessary to treat one, or perhaps two, of theextracted components recursively as it dominates volume changes and defines the P-values forall minor components, e.g. in the high active first stage in the Purex process nitric acid anduranium define the flow rates and extraction factors for all other elements. In industrial processes it is not only important to extract the desired compound with high yieldand purity, but also to be able to strip it again with good efficiency using a minimum amountof chemicals. Contrary to single stage batch extraction, the possibility to vary the P-value bychanges in the flow rate ratio (remember that P = D × V /V ) makes the use of conditions
with D-values for the product not too far from 1 desirable. In such a case the product can bestripped by a decrease in the flow rate ratio (in more dilute form than in the feed) and amoderate decrease in the D-value without the need to introduce new chemicals for stripping. A typical example is the uranium purification part of the Purex process. Here uranium (andnitric acid) is extracted at high concentration in the presence of 1!6 M HNO and stripped at
a low flow rate ratio by more dilute nitric acid. If needed, the separation can be repeated afterreconcentration by evaporation. A.4. Solvent extraction equipment
The less conventional part ! from a chemical engineering viewpoint ! of a reprocessing plantis the solvent extraction equipment, even though the technique is becoming increasinglycommon in the chemical industry. The principle of all such equipment is illustrated by FigureA.1(a); it contains a mixing section for efficient transfer of materials between the phases anda settling section for efficient phase separation. The input and outputs provide for connectingstages in the countercurrent extraction scheme (Fig. A.2). Mixer-settlers (Fig. A.1(a)) providegood mixing and reasonably good phase separation performance but rather large hold-ups. For reprocessing the equipment must be highly reliable, have high stage efficiency, shortcontact times, small liquid inventory (hold-up), be easy to decontaminate and to service, andnot least be safe against criticality. High reliability usually means simple design and few (if any)moving parts. The packed columns meet this requirement. They are simply long columns (often10!20 m with a diameter of 0.3!3 m) filled with small pieces of material obstructing a straightflow through the column, which is by gravity (Fig. A.1(b)).
However, high stage efficiency requires mechanical agitation of the two phases and clean phaseseparation, which cannot be met by packed columns. In pulsed columns (Fig. A.1(c)) the mechanical agitation provides good mixing but poor phaseseparation. Each plate is perforated (a sieve). The organic and aqueous phases separate betweenthe plates ("settling chambers"). In the down movement (<3 of the interplate distance) theaqueous phase is forced through the sieves, forming droplets, which by gravity fall through thelighter organic phase and coalesce when reaching the interface boundary. In the upward stroke,organic droplets form and rise through the aqueous phase until they meet the organic phaseboundary. The phase separation in a mixer-settler battery or in a column is usually not better than~99%, i.e. each outgoing phase contains some percent entrained droplets of the other phase. This separation efficiency can be improved to almost 100% by using centrifugal extractors (Fig. A.1(d)). Centrifugal extractors effect good mixing, good phase separation, and have very smallhold-ups. The organic-aqueous phase contact time in centrifugal extractors can be made muchshorter than in mixer-settlers or columns. The small hold-up volume and short residence timecooperate to reduce radiation decomposition. Packed columns were used in the first Windscale plant (Sellafield, UK). Pulsed columns wereused at Hanford (USA), in the old Eurochemic plant at Mol (Belgium), and are currently in usein the newer La Hague and THORP plants. Mixer-settlers have been used at Savannah River(USA), in the Magnox plant at Sellafield (UK), and at La Hague (France). Centrifugalextractors have been installed at Savannah River and at La Hague. A.5. Exercises A.1. In a solvent system the distribution ratio, D , is 2 for uranium and D is 0.003 for cesium. If 99.5% U is to be
extracted in a repeated batch fashion (eqn. (A.5)), (a) how much Cs is coextracted? If instead a countercurrent processis used with 10 extraction and 2 wash stages, what percentage of (b) uranium and (c) cesium is extracted? In an extractionequipment D 0.003 cannot be maintained, because droplets are carried over between stages; the practical value will be
D' 0.02. (d) How much cesium is extracted in this latter case with the countercurrent equipment? Assume equal phase
volumes. A.2. After scrubbing the solvent (1M TBP in kerosene) with a carbonate solution it should be reacidified to equilibrium with 6 M nitric acid before reuse in reprocessing operations. Assume that the partial molar volume of nitric acid can be neglected for both the organic phase and the aqueous phase. Thus, in this example extraction of nitric acid will not increase the volume of the organic phase nor reduce the volume of the aqueous phase. Extraction of nitric acid is assumed to only occur as the complex TBP.HNO3. For simplicity we will assume that the equation D
, with k = 0.1 can be used to calculate the D-value for nitric acid. The reacidification will
be performed in a simple counter current mixer-settler battery. (a) How many stages and (b) what flow rate ratio (org/aq)would be needed in order to produce the required nitric acid concentration in the organic phase using the smallest possiblefeed of 6.4 M nitric acid? (c) What would be the concentration of the spent nitric acid?
A.6. Literature
G. H. MORRISON and H. FREISER, Solvent Extraction in Analytical Chemistry, Wiley, 1957. Y. MARCUS and A. S. KERTES, Ion Exchange and Solvent Extraction of Metal Complexes, Wiley, 1969. A. K. DE, S. M. KHOPKAR and R. A. CHALMERS, Solvent Extraction of Metals, Van Nostrand Reinhold, 1970. YU. A. ZOLOTOV, Extraction of Chelate Complexes, Humprey Sci. Publ., Ann Arbor, 1970. T. SEKINE and Y. HASEGAWA, Solvent Extraction Chemistry, Marcel Dekker, 1977. T. C. LO, M. H. I. BAIRD and C. HANSON, Handbook of Solvent Extraction, Wiley, 1983. Radiochemistry and Nuclear Chemistry
G. RITCEY and A. W. ASHBROOK, Solvet Extraction. Part I and II, Elsevier, 1984. J. RYDBERG, C. MUSIKAS and G. CHOPPIN, Principles and Practices of Solvent Extraction, Marcel Dekker, 1992. (Journal of) Solvent Extraction and Ion Exchange, Marcel Dekker (Vol. 11, 1993).
Clarithromycin-Resistant Helicobacter Pylori Strains among Dyspeptic Patients in Sudan Nazar Abdalazeem*1, Hassan Abdul-Aziz1, Adam Ahmed Adam2, Waleed Hussein Omer2 and *1Ahfad University for Women, Omdurman, Sudan 1Alribat University, Academic Affairs 2Al-Neelain University, Faculty of Medicine and Al-Neelain Medical Research Centre, Sudan Corresponding author: P.O.Box:167, Omdurman,
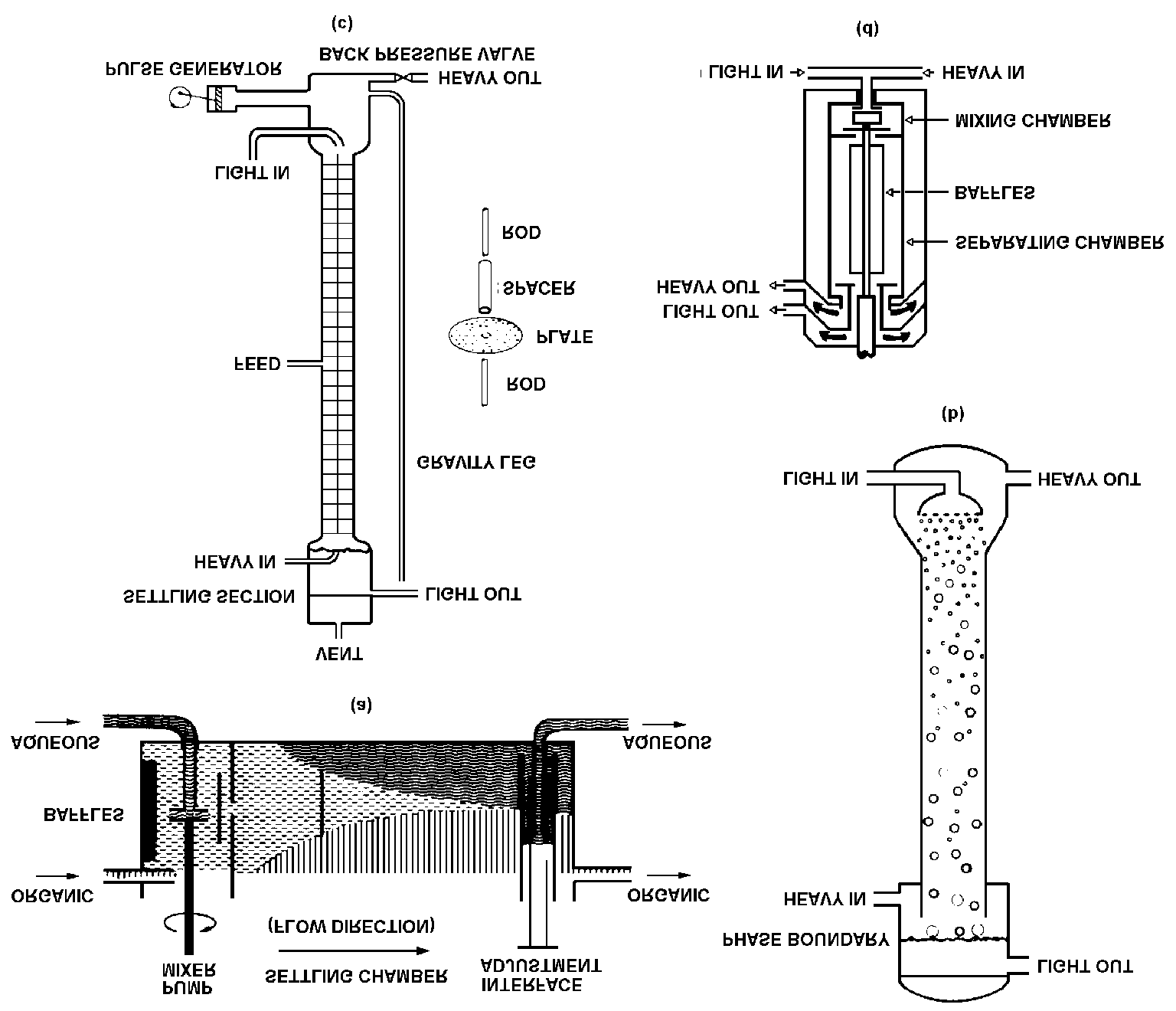 FIG. A.1. Different types of continuous extraction equipment. (a) Mixer-settler. (b) Spray column. (c) Pulsed column.
FIG. A.1. Different types of continuous extraction equipment. (a) Mixer-settler. (b) Spray column. (c) Pulsed column.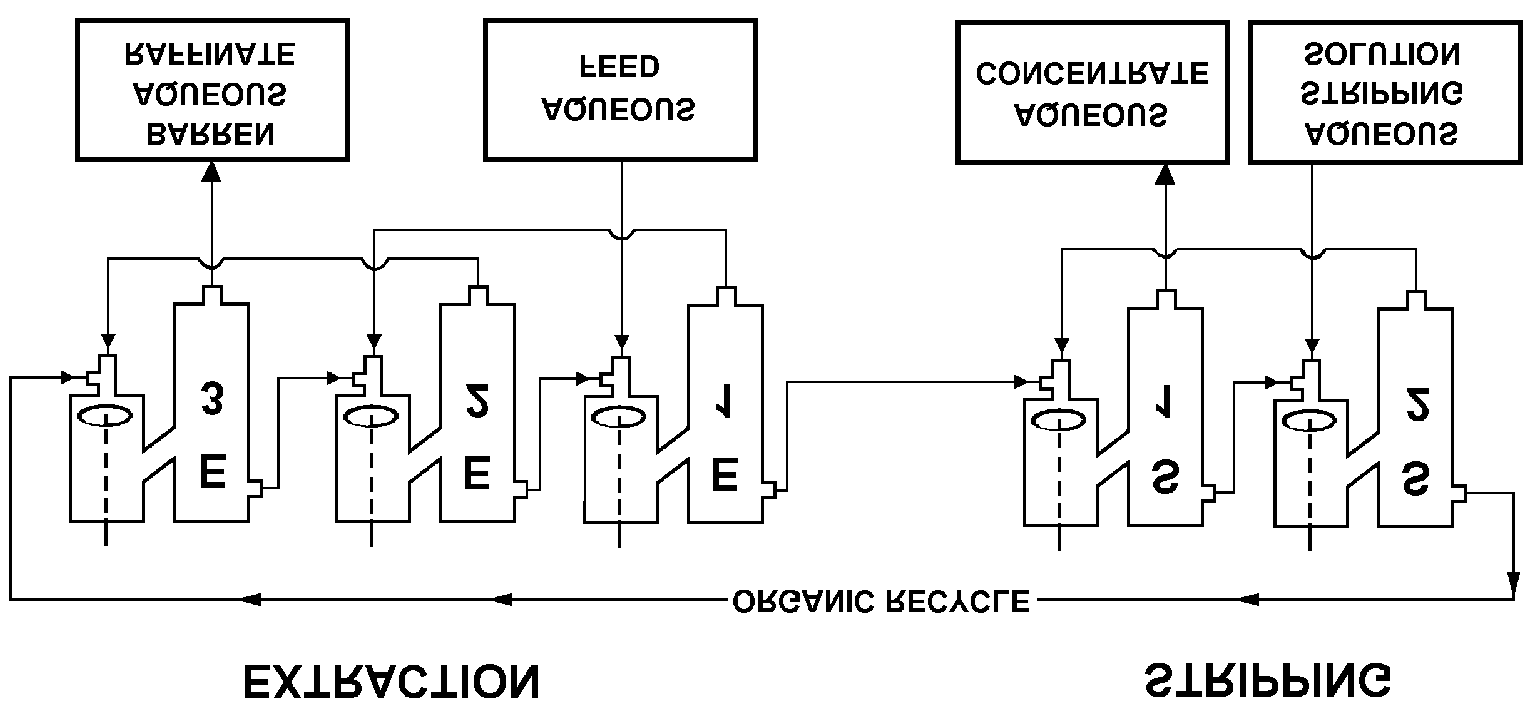
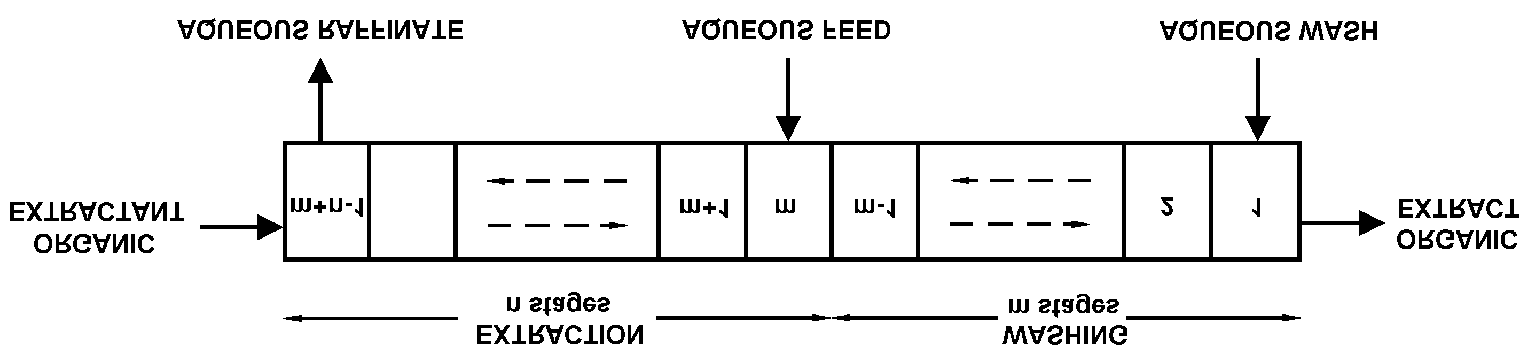 FIG. A.2. Mixer-settler countercurrent solvent extraction battery.
FIG. A.2. Mixer-settler countercurrent solvent extraction battery.