J. Nat. Prod. 2005, 68, 1071-1075 Phenanthroindolizidine Alkaloids from the Stems of Ficus septica
Amooru G. Damu,† Ping-Chung Kuo,†,| Li-Shian Shi,†,| Chia-Ying Li,† Chang-Sheng Kuoh,‡ Pei-Lin Wu,† andTian-Shung Wu*,†,§
Department of Chemistry, National Cheng Kung University, Tainan, Taiwan, Republic of China, Department of Life Science,National Cheng Kung University, Tainan, Taiwan, Republic of China, and National Research Institute of Chinese Medicine,Taipei, Taiwan, Republic of China
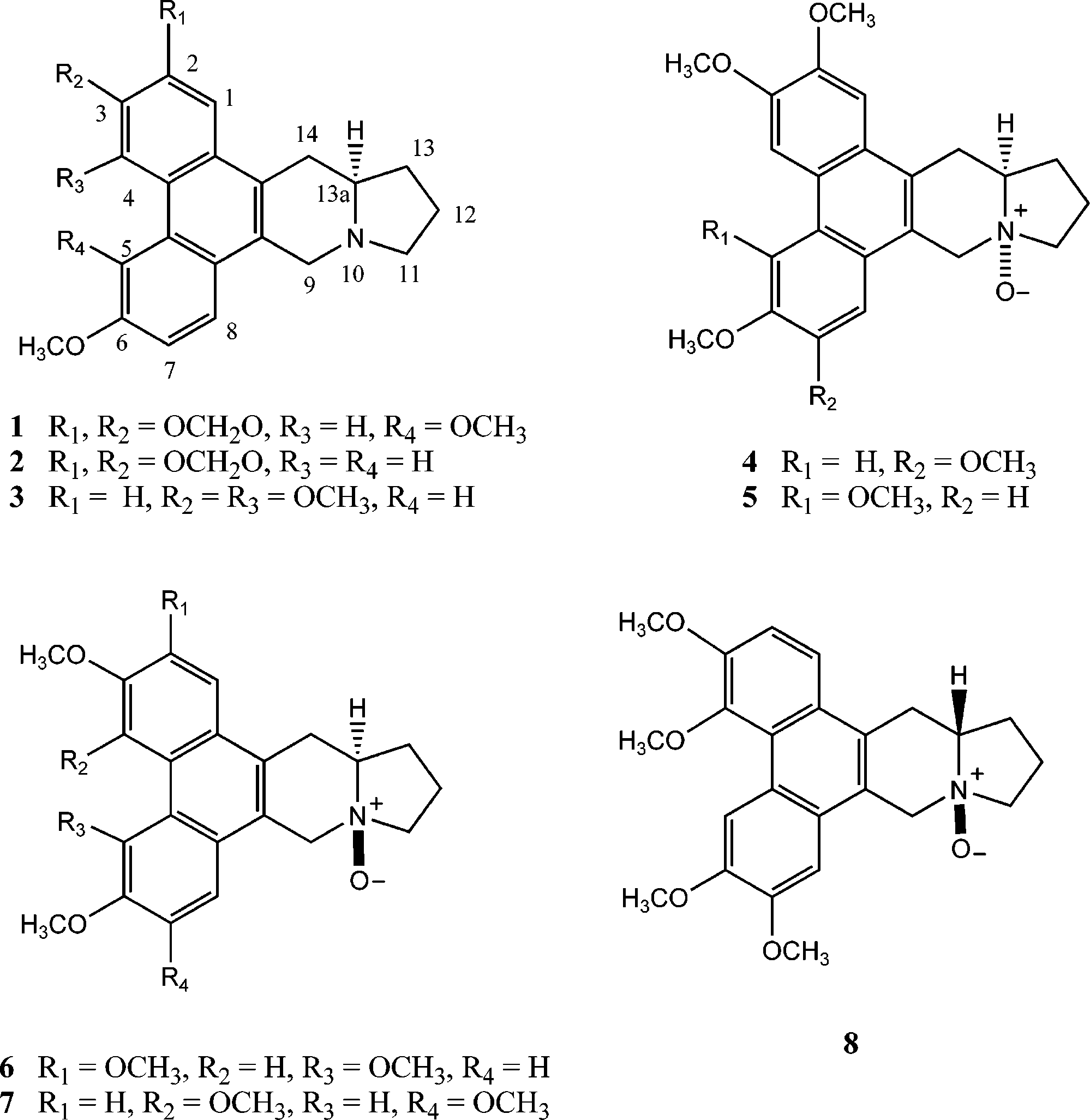
In addition to six known phenanthroindolizidine alkaloids, eight new alkaloids, namely, ficuseptines B-D (1-3), 10R,13aR-tylophorine N-oxide (4), 10R,13aR-tylocrebrine N-oxide (5), 10S,13aR-tylocrebrine N-oxide (6), 10S,13aR-isotylocrebrine N-oxide (7), and 10S,13aS-isotylocrebrine N-oxide (8), were isolated from a methanol extract of the stems of Ficus septica. The structures of the new compounds were elucidated by means of spectroscopic data interpretation. Cytotoxicity of some of these alkaloids was assessed in vitro using the HONE-1 and NUGC cell lines. Ficus septica Burm. f. (Moraceae) is a subtropical tree,
which occurs widely in low-altitude forests of Taiwan.1 This species has been known for its detoxicant, purgative, and emetic effects.1 The leaves of this plant have been used in folk medicine to treat colds, fever, and fungal and bacterial diseases.2-4 Several phenanthroindolizidine alkaloids, tri- terpenoids, lignans, acetophenones, steroids, and long- chain aliphatic compounds have been reported earlier from the leaves and roots of F. septica.5-10 Members of the phenanthroindolizidine alkaloid class are known to exhibit pronounced cytotoxicity and antiamebic, antifungal, anti- bacterial, and antiinflammatory activities and to also inhibit enzymes involved in the synthesis of DNA and proteins.7,11-25 In our ongoing investigations on cytotoxic constituents from plants native to Taiwan, it was found that the methanol extract of the stem of F. septica exhibited potent cytotoxicity against the HONE-1 and NUGC cell lines. Purification of the alkaloidal fractions of the metha- nol extract by eluting over silica gel followed by HPLC afforded eight new phenanthroindolizidine alkaloids (1- 8), along with six known analogues. In this paper, we report the isolation and characterization of these new phenan- throindolizidine alkaloids, which were found only as trace constituents of F. septica stems. Results and Discussion
consistent with the molecular formula C23H23NO4. Its UV
Bioassay-guided fractionation and separation of metha-
spectrum showed maxima at 255, 261, 281, 340, and 358
nolic extract of the stems of F. septica and subsequent
nm, indicating a substituted phenanthrene ring system.28
HPLC purification gave eight new phenanthroindolizidine
A set of ortho-coupled doublets (J ) 9.2 Hz) at δ 7.42 and
alkaloids (1-8), together with six known compounds,
7.71 in 1H NMR spectrum was assigned to H-7 and H-8,
tylophorine,26 tylocrebrine,5 isotylocrebrine,26 dehydroty-
respectively, since the latter proton showed a NOE cor-
lophorine,27 10S,13aR-tylophorine N-oxide,26 and 10S,-
relation with H-9 at δ 4.58. The two aromatic singlets at
13aR-antofine N-oxide.16 Although a comparatively large
δ 9.18 and 7.45 were attributed to para positions, H-4 and
quantity of plant material was collected for this investiga-
H-1, respectively, of ring A, based on the NOE interaction
tion, these labile alkaloids were obtained in very low yields.
between H-1 and H-14 (δ 3.28). The 1H NMR spectrum
Accordingly, it was not possible to obtain 13C NMR spec-
of 1 also displayed signals corresponding to two methoxyl
troscopic data to aid in their structure elucidation in the
groups at δ 3.88 (3H, s) and 4.01 (3H, s) and one set of
methylenedioxy protons at δ 6.15 (2H, s). An intense
Ficuseptine B (1), obtained as a colorless gum, showed
fragment ion peak was observed at m/z 308 in the EIMS,
a molecular ion peak at m/z 377.1630 in its HREIMS,
suggesting that these two methoxyls and the methylene-dioxy group are present in the phenanthrene moiety. The
* To whom correspondence should be addressed. Tel: 886-6-2747538.
placement of one of the methoxyl groups on C-6 was
Fax: 886-6-2740552. E-mail: tswu@mail.ncku.edu.tw.
supported by the NOE cross-peak between H-7 and a
Department of Chemistry, National Cheng Kung University.
Department of Biotechnology, National Formosa
methoxyl at δ 4.01. On the other hand, the methoxyl at δ
University, Yunlin, Taiwan, Republic of China.
3.88 showed a NOE correlation between H-4 and OCH3-6,
Department of Life Science, National Cheng Kung University.
§ National Research Institute of Chinese Medicine.
suggesting that it is attached to C-5. Accordingly, the
2005 American Chemical Society and American Society of Pharmacognosy
Journal of Natural Products, 2005, Vol. 68, No. 7Table 1. 1H NMR Data of Compounds 1-8
methylenedioxy group was placed at C-2 and C-3. The
tions of H-1/H-14 (δ 3.35) and H-4/H-5. The stereochem-
absolute stereochemistry of 1 was determined from the
istry of C-13a was determined as having the R configura-
optical rotation and CD spectrum. Thus, phenanthroin-
tion on the basis of the negative optical rotation value and
dolizidine alkaloids with the R configuration at C-13a have
a positive Cotton effect at 257 nm in the CD spectrum, and
been found to exhibit a negative optical rotation and a
thus H-13a was R oriented.29,30 Finally, the NOE between
positive Cotton effect around 260 nm.29,30 Compound 1 was
H-13a and H-9R indicated the existence of a chair-like
in agreement with this, as it showed a negative optical
conformation for the piperidine ring.31 Thus, the structure
rotation measured at the sodium-D line and a positive
of 2 was elucidated as 6-methoxy-2,3-methylenedioxy-
Cotton effect at 259 nm in the CD spectrum. Thus, 1
13aR-phenanthroindolizidine, and it has been designated
possesses the R configuration at C-13a. The presence of a
NOE between H-13a and H-9R suggested that the piperi-
Ficuseptine D (3) exhibited a molecular formula of
dine ring adopts a chair-like conformation.31 On the basis
C23H25NO3, based on a molecular ion peak at m/z 363.1833
of the foregoing spectroscopic studies, the structure of
in its HREIMS. Its UV absorption maxima at 252, 260, 280,
compound 1 was established as 5,6-dimethoxy-2,3-meth-
342, and 351 nm suggested it to be a substituted phenan-
ylenedioxy-13aR-phenanthroindolizidine, for which the
threne derivative.28 The 1H NMR spectrum of 3 displayed
trivial name ficuseptine B was proposed, following a
signals for three methoxyl groups at δ 3.92, 3.98, and 4.05
previous convention for these alkaloidal constituents of F.
(each 3H, s). Since 3 showed a similar pattern of 1H NMR
signals due to the indolizidine moiety as observed for 2
Ficuseptine C (2) was obtained as colorless gum, and its
(Table 1), three methoxyl groups could be assigned in the
UV absorption maxima were observed at 257, 282, and 340
phenanthrene moiety. This conclusion was supported by
nm, very similar to those of trioxygenated phenanthrene
an intense fragment ion peak at m/z 294 in the EIMS. The
derivatives.28 It exhibited a molecular ion peak at m/zortho-coupled doublets appearing at δ 7.49 and 7.83 (each
347.3513 in its HRFABMS, corresponding to the molecular
1H, d, J ) 8.4 Hz) were assigned to H-1 and H-2 of ring A,
formula C22H21NO3, 30 amu less than that of 1. The
as a NOE correlation was observed between H-1 and H-14
observation of an intense fragment ion at m/z 278 in the
(δ 3.32). A set of ABX pattern signals at δ 9.31 (1H, d, J )
EIMS due to a phenanthrene moiety suggested the pres-
2.8 Hz), 7.25 (1H, dd, J ) 7.2, 2.8 Hz), and 7.87 (1H, d, J
ence of one methoxyl group and one methylenedioxy group,
) 7.2 Hz) were attributed to H-5, H-7, and H-8 of ring B
instead of two methoxyl groups and one methylenedioxy
on the basis of the NOEs of H-8 with H-9R (δ 4.58) and
group as in 1. As expected, the 1H NMR spectrum also
H-7. The NOE correlation of the methoxyl protons at δ 3.92
displayed signals for methoxyl protons at δ 4.02 (3H, s)
with H-7 and H-5 was used to determine the location of
and methylenedioxy protons at δ 6.15 (2H, s). Since the
the methoxyl group at C-6. The remaining two methoxyls
proton signals of the B ring appeared as an ABX pattern
at δ 3.98 and 4.05 were placed at C-3 and C-4, respectively,
at δ 8.05 (1H, d, J ) 2.8 Hz, H-5), 7.86 (1H, d, J ) 8.8 Hz,
as the former gave a NOE with H-2 and the latter with
H-8), and 7.21 (1H, dd, J ) 8.8, 2.8 Hz, H-7), and NOEs of
H-5. The negative optical rotation value and positive Cotton
H-8/H-9R (δ 4.65), H-5/H-4 (δ 8.18) and the methoxyl
effect at 259 nm in the CD spectrum of 3 supported the R
protons/H-5 and H-7 were observed, the methoxyl group
configuration at C-13a.29,30 In addition, a NOE correlation
was placed at C-6. The attachment of the methylenedioxy
between H-13a and H-9R suggested a chair-like conforma-
group at C-2 and C-3 was confirmed by the multiplicities
tion for the piperidine ring.31 Accordingly, the structure of
for H-1 and H-4 of ring A as two aromatic singlets at δ3 (ficuseptine D) was concluded to be 3,4,6-trimethoxy-
7.43 (1H, s, H-1) and 8.18 (1H, s, H-4) and NOE correla-
Phenanthroindolizidine Alkaloids from FicusJournal of Natural Products, 2005, Vol. 68, No. 7
Alkaloid 4, obtained as yellow needles, was considered
OCH3-6; and H-8/H-7, H-9R. The positive CD curve at 261
to have the same molecular formula, C24H27NO5, as tylo-
nm and a negative optical rotation value inferred the R
phorine N-oxide, on the basis of its HREIMS, suggesting
stereochemistry at C-13a and thus H-13a to be in an R
it to be an isomer.15 The 1H NMR spectrum revealed the
orientation.29,30 The resonance due to H-13a at δ 3.45 was
presence of four methoxyl group signals at δ 4.02, 4.04
indicative of a trans-fused indolizidine ring junction.32,33
(each 3H, s) and 4.09 (6H, s). Since the UV spectroscopic
pattern and 1H NMR signals and coupling patterns corre-
(δ 3.65) by oxygen led to the assignment of the
sponding to the phenanthrene moiety were similar to those
configuration of the N-oxide group.16,33 Finally, the obser-
of tylophorine N-oxide,15 the four methoxyl groups could
vation of a NOE between H-13a and H-9R indicated that
be located at C-2, C-3, C-6, and C-7. This was supported
the piperidine ring adopted a chair-like conformation as
by NOE cross-peaks between H-1/H-14R, H-14 , OCH3-2;
in 4.31 Consequently, 6 was identified as 10S,13aR-tylo-
H-4/H-5, OCH3-3; OCH3-2/OCH3-3; OCH3-6/H-5, OCH3-7;
and H-8/H-9R, H-9 , OCH3-7. Evidence for the presence of
Alkaloid 7 was obtained as pale yellow needles. On the
an N-oxide unit was also suggested by the downfield shifts
basis of the HREIMS data 7 was suggested to have the
of H-9R (δ 5.17), H-9 (δ 4.86), H-11R (δ 3.80), H-11 (δ
same molecular formula, C24H27NO5, as isotylocrebrine
3.72), and H-13a (δ 3.94) and by the prominent [M - 16]+
N-oxide,15 indicating these alkaloids to be isomers. Accord-
peak at m/z 393 in the mass spectrum of 4. The negative
ingly, two mutually coupled doublets (J ) 9.2 Hz) at δ 7.85
optical rotation measured at the sodium-D line and positive
and 7.45 and two singlets at δ 9.30 and 6.98 in its 1H NMR
Cotton effect at 256 nm in the CD spectrum for 4 inferred
spectrum were assigned to H-1, H-2, H-5, and H-8, respec-
the C-13a R configuration.29,30 Thus, H-13a was oriented
tively.15 Thus, four methoxyl groups at δ 4.02, 4.01, 3.92,
in an R direction. The H-13a proton resonated at δ 3.94
and 3.99 could be placed, in turn, at C-3, C-4, C-6, and C-7.
and indicated a cis-fused ring junction of the indolizidine
These assignments were confirmed by the correlations of
moiety.32,33 Moreover, the strong deshielding of H-9R (δ
H-1/H-14 ; H-2/OCH3-3; H-5/OCH3-4, OCH3-6; and H-8/
5.17) and H-11R (δ 3.80) by oxygen supported the R
OCH3-7, H-9R in a NOESY experiment. The positive Cotton
configuration of the N-oxide group.16,33 The observation of
effect at 279 nm in the CD spectrum of 7 and negative
NOE correlations between H-1 and H-14R, H-14 ; H-8 and
optical rotation inferred the R configuration at C-13a.29,30
H-9R, H-9 ; H-9R and H-11R; and H-14R and H-13R in the
Hence, H-13a was located with an R orientation. The
NOESY spectrum suggested that the indolizidine moiety
appearance of the H-13a signal at δ 3.51 led the ring
adopted a boat-like conformation.31 Hence, 4 was charac-
junction configuration to be determined as trans32,33 for the
terized as 10R,13aR-tylophorine N-oxide.
indolizidine unit. Moreover, the strong deshielding of H-9
Alkaloid 5 was assigned a molecular formula of C δ 4.65) and H-11 (δ 3.68) by oxygen also inferred the
configuration of the N-oxide group.16,33 The appearance of
5 from its HREIMS, one oxygen atom more than that
of tylocrebrine, suggesting the presence of an N-oxide
NOE cross-peaks between H-13a and H-9R was indicative
functionality. 1H NMR multiplicities of the four aromatic
of a chair-like conformation for the piperidine ring.31 The
proton signals and four methoxyl groups similar to those
structure of 7 was, therefore, defined as 10S,13aR-iso-
of tylocrebrine, and the fragment ion peak at m/z 324, due
to the phenanthrene moiety in EIMS, allowed the four
Alkaloid 8 was obtained as pale yellow needles. On the
methoxyl groups to be fixed at C-2, C-3, C-5, and C-6,
basis of the molecular formula, C24H27NO5, as determined
respectively. This was further supported by NOEs of H-1/
from the HREIMS, 8 was considered to be an isomer of 7.
H-14R, H-14 , OCH3-2; H-4/OCH3-3; OCH3-5, OCH3-6/H-
Its UV spectrum showed absorptions at 245, 264, 285, 344,
7; and H-8/H-7, H-9R, H-9 . These assignments indicated
and 360 nm and was practically superimposable on that
that the extra oxygen must be present in the indolizidine
of 7. In the 1H NMR spectrum, four methoxyls and four
moiety. Evidence for the presence of an N-oxide unit was
aromatic proton signals were observed with the same
also revealed by the significant [M - 16]+ ion peak at m/z
multiplicities as those of 7, but differed in their indolizidine
393 and downfield shifts of H-9R (δ 5.13), H-9 (δ 4.81),
protons (Table 1). The placement of four methoxyl groups
H-11R (δ 3.72), H-11 (δ 3.65), and H-13a (δ 3.98) in the
at C-3, C-4, C-6, and C-7 was determined from NOEs
1H NMR spectrum. The negative optical rotation value and
between H-1/H-14 ; H-2/OCH3-3; H-5/OCH3-4, OCH3-6;
positive Cotton effect at 275 nm in the CD spectrum
and H-8/OCH3-7, H-9R. The positive specific rotation and
established the R stereochemistry at C-13a, with H-13a in
positive Cotton effect at 279 nm in the CD spectrum of 8
an R orientation.29,30 A strong deshielding of H-13a to δ
confirmed the S configuration of C-13a16,26 and the
3.98 inferred a cis-fused ring junction of the indolizidine
orientation of H-13a. The downfield shift of H-13a to δ 4.08
moiety.32,33 The R orientation of the N-oxide group was also
inferred that the indolizidine ring junction should be in the
indicated by a strong deshielding of H-9R and H-11R by
cis32,33 configuration. Furthermore, the
the oxygen atom.16,33 Similar NOE correlations between
the N-oxide group was also inferred from a strong deshield-
H-1 and H-14R, H-14 ; H-8 and H-9R, H-9 ; H-9R and
H-11R; and H-14R and H-13R as in 4 suggested a boat-
appearance of NOE correlations between H-1 and H-14R,
like conformation for the indolizidine moiety.31 Thus, 5 was
H-14 ; H-8 and H-9R, H-9 ; H-9R and H-11R; and H-14R
concluded to be 10R,13aR-tylocrebrine N-oxide.
and H-13R in the NOESY spectrum indicated that the
Alkaloid 6 was isolated as pale yellow needles. The
indolizidine moiety adopted a boat-like conformation.31
Therefore, it was concluded that the 8 is 10S,13aS-
IMS, was the same as that of 5, indicating these alkaloids
to be isomers. On the basis of the fact that 6 showed four
Compounds 6, 7, and tylophorine were tested in vitro
aromatic proton signals and four methoxyl signals in the
for their cytotoxicity using HONE-1 and NUGC tumor cell
1H NMR spectrum similar to those of 5 (Table 1), the
lines.9,34 All three compounds exhibited strong cytotoxicity
substituents were assigned to C-2, C-3, C-5, and C-6 as in
against both HONE-1 and NUGC cell lines. The percent-
5. These assignments were supported by NOESY correla-
ages of inhibition observed for 6, 7, and tylophorine at 10
tions of H-1/H-14 , OCH3-2; H-4/OCH3-3, OCH3-5; H-7/
µM concentration against HONE-1 cell lines were 92%,
Journal of Natural Products, 2005, Vol. 68, No. 7
87%, and 80%, respectively, whereas against NUGC cell
1711, 1683, 1511, 1467, 1424, 1396, 1286, 1010 cm-1; CD (c
lines they exhibited 94%, 93%, and 85% inhibition, respec-
tively, at the same concentration. Compound quantities
MHz), see Table 1; EIMS m/z 377 [M]+ (43), 308 (100), 293
available did not permit the more formal determination of
(34), 277 (15), 129 (10), 96 (13), 69 (20); HREIMS m/z 377.1630([M]+) (calcd for C
Ficuseptine C (2): colorless gum; [R] 25 - Experimental Section
MeOH); UV (MeOH) λmax (log ) 257 (3.46), 282 (3.42), 340(2.68) nm; IR (KBr) νmax 2925, 2853, 2375, 2311, 1715, 1710,
General Experimental Procedures. Optical rotations
1683, 1548, 1514, 1454, 1412, 1391, 1371, 1212, 1035 cm-1;
were measured using a JASCO DIP-370 digital polarimeter.
CD (c 1.09 × 10-4, MeOH) [θ]
Circular dichroism (CD) and UV spectra were recorded at room
400 MHz), see Table 1; EIMS m/z 347 [M]+ (31), 278 (100),
temperature on a JASCO J-700 spectropolarimeter and on a
264 (10), 248 (29), 175 (11), 153 (10), 129 (20), 95 (11), 69 (65);
Hitachi UV-3210 spectrophotometer, respectively. IR spectra
HREIMS m/z 347.3513 ([M]+) (calcd for C22H21NO3, 347.3515).
were obtained with a Shimadzu FT-IR DR-8011 spectropho-
Ficuseptine D (3): colorless gum; [R] 25 -
tometer. NMR spectra were recorded on Bruker AMX-400,
MeOH); UV (MeOH) λmax (log ) 252 (sh) (3.58), 260 (3.62), 280
AVANCE-300, and Varian Unity Plus 400 spectrometers.
(2.68), 342 (2.61), 351 (3.21) nm; IR (KBr) νmax 2923, 1679,
Chemical shifts are shown in δ values (ppm) with tetrameth-
1668, 1659, 1600, 1514, 1455, 1410, 1398, 1282, 1010 cm-1;
ylsilane as an internal standard. Mass spectra were measured
CD (c 1.40 × 10-5, MeOH) [θ]259 3177; 1H NMR (acetone-d6,
on a VG-70-250S spectrometer with EI or FAB ionization
400 MHz), see Table 1; EIMS m/z 363 [M]+ (40), 294 (100),
(positive-ion mode). Column chromatography was performed
279 (18), 236 (10), 129 (11), 97 (15), 69 (21); HREIMS m/z363.1833 ([M]+) (calcd for C
on silica gel (70-230 mesh, 230-400 mesh). Fractions were
10R,13aR-Tylophorine N-oxide (4): pale yellow needles;
monitored by TLC (Merck precoated Si gel 60 F254 plates),
mp 210-220 °C (dec); [R] 25 -77.7° (c 0.019, MeOH); UV
using UV light and Dragendorff’s reagent to visualize the
(MeOH) λmax (log ) 241 (4.09), 258 (4.13), 287 (3.84), 303 (3.56),
spots. High-performance liquid chromatography (HPLC) was
340 (2.52) nm; IR (KBr) νmax 2925, 1741, 1691, 1647, 1515,
performed on a Shimadzu LC-10ATVP series pumping system
1463, 1427, 1315, 1257, 1153, 1039 cm-1; CD (c 4.55 × 10-5,
equipped with a Shimadzu SPD-6AV UV-vis spectrophoto-
metric detector at 275 nm, a Cosmosil packed column with
1; EIMS m/z 409 [M]+ (5), 393 (20), 392 (8), 337 (74), 319 (54),
5C18-AR-II Waters type (4.6 × 250 mm, 5 µm) and a
176 (32), 154 (100), 136 (76), 86 (20); HRFABMS m/z 410.4748
Lichrospher 100 RP-8 column (4.6 × 250 mm, 5 µm), and a
([M + H]+) (calcd for C24H28NO5, 410.4749). 10R,13aR-Tylocrebrine N-oxide (5): pale yellow needles; Plant Material. The stems of F. septica were collected in
Tainan Hsien, Taiwan, Republic of China, in January 2000
(MeOH) λmax (log ) 244 (4.14), 265 (4.29), 276 (3.71), 310 (3.46)
and were authenticated by Prof. C. S. Kuoh, Department of
320 (3.41) nm; IR (KBr) νmax 2941, 1741, 1706, 1562, 1515,
Life Science, National Cheng Kung University. A voucher
1477, 1463, 1427, 1398, 1365, 1284, 1253, 1114, 1068, 1026
specimen (Wu 200000053) has been deposited in the Her-
cm-1; CD (c 6.36 × 10-5, MeOH) [θ]
barium of National Cheng Kung University, Tainan, Taiwan.
OD, 400 MHz), see Table 1; EIMS m/z 409 [M]+ (8), 393 (48),
Extraction and Isolation. Dried and powdered stem of
392 (24), 337 (100), 324 (99), 176 (9), 154 (21), 86 (10);
F. septica (46.5 kg) was refluxed with methanol (7 × 10 L)
HRFABMS m/z 410.3916 ([M + H]+) (calcd for C24H28NO5,
and filtered. A large amount of precipitate, asparagine (260
g),35 was formed during this process and filtered. The filtrate
10S,13aR-Tylocrebrine N-oxide (6): pale yellow needles;
was concentrated and suspended in water. Then the water
solubles were partitioned with chloroform and n-butanol,
(MeOH) λmax (log ) 245 (3.14), 264 (4.39), 279 (3.91), 285 (3.82),
successively. The concentrated chloroform extract (80 g) was
307 (3.41), 319 (3.38) nm; IR (KBr) νmax 2937, 1741, 1606, 1515,
subjected to open column chromatography over silica gel by
1463, 1396, 1286, 1255, 1211, 1114, 1035, 1026 cm-1; CD (c
eluting with a stepwise gradient from 5% to 75% methanol in
chloroform to afford 12 fractions. Fractions were monitored
see Table 1; EIMS m/z 409 [M]+ (24), 393 (80), 337 (10), 324
by TLC using Dragendorff’s reagent to visualize the spots and
(100), 154 (61), 136 (46), 86 (14); HRFABMS m/z 410.4416
tested for their cytotoxicity. Fractions 5-9 were considered to
([M + H]+) (calcd for C24H28NO5, 410.4413).
be active fractions, since they showed cytotoxicity against the
10S,13aR-Isotylocrebrine N-oxide (7): pale yellow needles;
HONE-1 and NUGC cell lines with the inhibition percentage
values of 93% and 97%, 80% and 90%, 79% and 92%, 75% and
(MeOH) λmax (log ) 245 (3.24), 263 (5.39), 283 (3.12), 287 (3.08),
84%, and 85% and 96%, respectively at 250 µM concentration.
309 (2.41) 344 (2.24), 359 (2.19) nm; IR (KBr) νmax 2943, 2842,
Fraction 5 on repeated column chromatography over silica gel
1691, 1635, 1521, 1458, 1413, 1267, 1114, 1028 cm-1; CD (c
and further purification by HPLC using 1 mL/min of MeOH-
H2O-Et2NH (75:24.5:0.5) gave compounds 1 (1.0 mg, 0.0012%),
see Table 1; EIMS m/z 409 [M]+ (14), 393 (80), 324 (100), 307
2 (1.0 mg, 0.0012%), 3 (1.0 mg, 0.0012%), 4 (7.0 mg, 0.0087%),
(10),176 (25), 154 (61), 136 (46), 86 (14); HRFABMS m/z
tylophorine (18.0 mg, 0.022%), tylocrebrine (1.0 mg, 0.0012%),
410.4961 ([M + H]+) (calcd for C24H28NO5, 410.4963).
isotylocrebrine (1.1 mg, 0.0013%), dehydrotylophorine (0.5 mg,
10S,13aS-Isotylocrebrine N-oxide (8): pale yellow needles;
0.00062%), and 10S,13aR-tylophorine N-oxide (1.5 mg, 0.0018%).
Similarly, repeated column chromatography and HPLC puri-
(MeOH) λmax (log ) 245 (3.21), 264 (5.41), 285 (3.17), 305 (2.89)
fication by using 1 mL/min of MeCN-H2O-Et2NH (20:79.5:
344 (2.21), 360 (2.14) nm; IR (KBr) νmax 2941, 2851, 1696, 1645,
0.5) of fraction 7 yielded compounds 6 (1.0 mg, 0.0012%) and
1562, 1463, 1255, 1114, 1024 cm-1; CD (c 5.01 × 10-5, MeOH)
8 (0.5 mg, 0.00062%). Fraction 8 was subjected to repeated
1128; 1H NMR (CD3OD, 400 MHz), see Table 1; EIMS
column chromatography separation and then HPLC purifica-
m/z 409 [M]+ (17), 393 (30), 324 (74), 307 (25),176 (12), 154
tion using 1 mL/min of MeCN-H2O-Et2NH (25:75:0.5) to
(100), 136 (74), 118 (66); HRFABMS m/z 410.3446 ([M + H]+)
obtain compounds 4 (1.9 mg, 0.0023%), 5 (2.6 mg, 0.0032%), 6
(2.6 mg, 0.0032%), 7 (5.9 mg, 0.0073%), 8 (2.5 mg, 0.0031%), Cytotoxicity Assay. Two human cancer cell lines, NUGC
and 10S,13aR-antofine N-oxide (2.5 mg, 0.0031%). An attempt
(gastric adenocarcinoma) and HONE-1 (nasopharyngeal car-
to work up cytotoxic fractions 6 and 9 did not lead to the
cinoma), were seeded in 96-well microliter plates at a density
isolation of any additional alkaloids due to the lability of these
of 6000/well in 10 µL of culture medium (Dulbecco’s modified
Eagle’s medium supplemented with 10% fetal calf serum and
Ficuseptine B (1): colorless gum; [R] 25 -
nonessential amino acid) and maintained at 37 °C in a
MeOH); UV (MeOH) λmax (log ) 255 (3.80), 261 (3.91), 281
humidified incubator with 5% CO2. After an overnight adapta-
(2.59), 340 (3.24), 358 (3.18) nm; IR (KBr) νmax 2924, 1745,
tion period, test compounds (final concentration, 50 µg/mL)
Phenanthroindolizidine Alkaloids from FicusJournal of Natural Products, 2005, Vol. 68, No. 7
in serum-free medium were added to individual wells. Cells
(14) Tanner, U.; Wiegrebe, W. Arch. Pharm. (Weinheim, Ger.) 1993, 326,
were treated with test compounds for 3 days. Cell viability
(15) Abe, F.; Hirokawa, M.; Yamauchi, T.; Honda, K.; Hayashi, N.; Ishii,
was determined by the 5-(3-carboxymethoxyphenyl)-2-(4,5-
M.; Imagawa, S.; Iwahana, M. Chem. Pharm. Bull. 1998, 46, 767-
dimethylthiazolyl)-3-(4-sulfophenyl)tetrazolium salt (MTS) re-
duction assay.9,32 The 5 µM (final concentration) actinomycin
(16) Staerk, D.; Christensen, J.; Lemmich, E.; Duus J.; Olsen, C. E.;
D (showed 100% of inhibition at 10 µM) and 0.3% (final
Jaroszewski, J. W. J. Nat. Prod. 2000, 63, 1584-1586.
concentration) DMSO were used as positive and vehicle
(17) Hartwell, J. L.; Abbot, B. J. In Advances in Pharmacology andChemotherapy; Schnitzer, R. J., Goldes, A., Eds.; Academic Press:
controls. Results were expressed as percent of DMSO control.
(18) Bhutani, K. K.; Sharma, G. L.; Ali, M. Planta Med. 1987, 47, 532- Acknowledgment. The authors are grateful for financial
support from the National Science Council, Taiwan, Republic
(19) Duan, J.; Yu, L. Zhongcaoyao 1991, 22, 316-318. (20) Narasimha Rao, K.; Bhattacharya, R. K.; Venkatachalam, S. R.
of China (NSC 93-2113-M-006-001), awarded to T.S.W. and
Cancer Lett. 1998, 128, 183-188.
also thankful to the National Research Institute of Chinese
(21) Narasimha Rao, K.; Venkatachalam, S. R. Toxicol. in Vitro 2000, 14,
Medicine, Taiwan, Republic of China, for partial financial
(22) Govindachari, T. R.; Viswanathan, N. Heterocycles 1978, 11, 587-
(23) Donaldson, G. R.; Atkinson, M. R.; Murray, A. W. Biochem. Biophys.References and Notes Res. Commun. 1968, 31, 104.
(24) Peraza-Sa´nchez, S. R.; Chai, H. B.; Shin, Y. G.; Santisuk, T.;
(1) Liao, J. C. Moraceae in Flora of Taiwan, 2nd ed.; Editorial Committee
Reutrakul, V.; Farnsworth, N. R.; Cordell, G. A.; Pezzuto, J. M.;
of the Flora of Taiwan: Taipei, 1996; Vol. 2, pp 177-180.
Kinghorn, A. D. Planta Med. 2002, 68, 186-188.
(2) Holdsworth, D. K.; Hurley, C. L.; Rayner, S. E. Q. J. Crude Drug
(25) Staerk, D.; Lykkeberg, A. K.; Christensen, J.; Budnik, B. A.; Abe, F.;
Res. 1980, 18, 131-139.
(3) Holdsworth, D.; Lacanienta, E. Q. J. Crude Drug Res. 1981, 19, 155-
Jaroszewski, J. W. J. Nat. Prod. 2002, 65, 1299-1302.
(26) Abe, F.; Uwase, Y.; Yamauchi, T.; Honda, K.; Hayashi, N. Phytochem-
(4) Holdsworth, D.; Damas, K. Int. J. Crude Drug Res. 1986, 24, 217- istry 1995, 39, 695-699.
(27) Govindachari, T. R.; Viswanathan, N.; Radhakrishnan, J.; Charubala,
(5) Russel, J. H. Naturwissenschaften 1963, 50, 443-444.
R.; Nityananda Rao, N.; Pai, B. R. Indian J. Chem. 1973, 11, 1215-
(6) Herbert, R. B.; Moody, C. J. Phytochemistry 1972, 11, 1184.
(7) Baumgartner, B.; Erdelmeier, C. A. J.; Wright, A. D.; Rali, T.; Sticher,
(28) Gellert, E. J. Nat. Prod. 1982, 45, 50-73.
O. Phytochemistry 1990, 29, 3327-3330.
(29) Mi, J. F.; Fang, S. D.; Chen, Y.; Xu, Y. M.; Zhang, R. Acta Pharm.
(8) Kuo, P. C.; Chiu, C. C.; Shi, S. L.; Li, C. Y.; Wu, S. J.; Damu, A. G.;
Sin. 1992, 27, 197-203.
Wu, P. L.; Kuoh, C. S.; Wu, T. S. J. Chin. Chem. Soc. 2002, 49, 113-
(30) Li, X.; Peng, J.; Onda, M. Heterocycles 1989, 29, 1797-1808.
(31) Ohmiya, S.; Kubo, H.; Nakaaze, Y.; Saito, K.; Murakoshi, I.; Otomasu,
(9) Wu, P. L.; Rao, K. V.; Su, C. H.; Kuoh, C. S.; Wu, T. S. Heterocycles
H. Chem. Pharm. Bull. 1991, 39, 1123-1125. 2002, 57, 2401-2408.
(32) Lavault, M.; Richomme, P.; Bruneton, J. Pharm. Acta Helv. 1994,
(10) Tsai, I. L.; Chen, J. H.; Duh, C. Y.; Chen, I. S. Chin. Pharm. J. 2000,
(33) Buckley, F. T.; Rapoport, H. J. Org. Chem. 1983, 48, 4222-4232.
(11) Gellert, E.; Rudats, R. J. Med. Chem. 1964, 7, 361-362.
(34) Cheng, M. J.; Lee, S. J.; Chang, Y. Y.; Wu, S. H.; Tsai, I. L.;
(12) Pettit, G. R.; Goswami, A.; Cragg, G. M.; Schmidt, J. M.; Zou, J. C.
Jayaprakasam, B.; Chen, I. S. Phytochemistry 2003, 63, 603-608. J. Nat. Prod. 1984, 47, 913-919.
(35) Wu, T. S.; Leu, Y. L.; Chan, Y. Y.; Lin, F. W.; Li, C. Y. Phytochemistry
(13) Suffness, M.; Cordell, G. A. In The Alkaloids: Chemistry and1998, 49, 1467-1470. Pharmacology; Brossi, A., Ed.; Academic Press: Orlando, 1985; Vol. 25, pp 1-355.
ABBEY MEAD SURGERY Practice ethos A forward-thinking, professional Practice – providing good quality, traditional, family medicine. APPOINTMENTS Providing enough appointments to suit everyone’s needs is a constant challenge! We need to have “BOOK ON THE DAY” appointments for people who have an acute or urgent problem and “BOOK IN ADVANCE” appointments (up to 4 weeks) f
CUSTOMS IN CONFLICT: Land Tenure Issues among Pastoralists in Ethiopia CUSTOMS IN CONFLICT: Land Tenure Issues among Pastoralists in Ethiopia ______________________________________________________________________ ABSTRACT This paper presents a broad review of land tenure issues among pastoral communities in the country. It is argued that the key element of pastoralism is that i
 J. Nat. Prod. 2005, 68, 1071-1075
J. Nat. Prod. 2005, 68, 1071-1075